Frequently asked questions
What are the sources included in FORENSIC?
To date, the fecal signature of 7 sources has been investigated for V4 and/or V6 regions of the 16S rRNA gene. It includes the human (sewage), dog, cow, deer (only v6), and pig source-classifiers, but also the ruminant (only v4) and/or the herbivore.
The source I would like to target is not in FORENSIC?
If you have enough representative samples, we could implement this source in FORENSIC. Let's talk about it.
What is a classifier?
A classifier contains the information of the fecal signature structure for a given source, i.e., the sequences and their relative abundance. The classifiers were created from 15-20 samples per source using sequences assigned to Bacteroidales or Clostridiales. More information about the classifiers can be found here.
What is the difference between global, draft and discarded classifiers?
We optimized the decision cut-off for each classifier to maximize their specificity (i.e., the proportion of negative samples correctly classified) and sensitivity (i.e., proportion of positive samples correctly classified) with a minimum percentgage of 70%. Because performances were evaluated using fecal samples and not environmental samples, having a voting decision cut-off too low can certainly increase sensitivity, but it can decrease the specificity leading to too many misclassifications. Classifiers with a cut-off ≤10% were considered a draft, while classifiers with a decision cut-off of ≤5% were discarded.
Why focus on Bacteroidales and Clostridiales bacterial orders?
Bacteroidales and Clostridiales are considered to be fecal-associated bacteria. Targeting these bacterial orders reduces the "noise" caused by environmental-associated bacteria. Moreover, Bacteroidales and Clostridiales are one of the predominant bacterial groups in mammals gut microbiota; their detection in environmental samples is, therefore, more likely. Last but not least, we consistently observe reliable patterns discriminating the different sources investigated, reflecting the host diet, physiology, etc. of the host.
My samples have a sequencing depth below the recommendations, can I still use FORENSIC?
Yes! However, depending on how shallow was the sequencing, it is likely that the fecal signature recovered from your samples will never be classified as "fecal signal detected (contaminated)". We evaluated that a sample with a fecal signature (i.e., matching the ASVs in the classifiers) represented by at least ~100 sequences is enough to generate accurate classifications.
I already processed my sequences using dada2, should I re-process everything using CUTADAPT and PEAR?
Preliminary explorations showed that FORENSIC could not correctly identify the bacterial signatures in tested samples using data processed by dada2. This incompatibility might originate from the fact that dada2 may shorten sequences of a nucleotide during the denoising step. This is not an issue when studying the whole bacterial community, but since FORENSIC is looking for the exact sequence matches between the signatures and the sample to test, predictions can be significantly altered.
How do I know my sequences are properly trimmed?
V4 and V6 classifiers are composed of sequences ranging from 250 to 254 nt, and from 54 to 66 nt, respectively. Because FORENSIC searchs for strictly identical sequences, shorter or longer sequences will cause FORENSIC to fail. If your sequences are too long, look for primers and trim your fasta using CUTADAPT.
Can I use data generated using another sequencing platform than Illumina?
We only validated the predictions using Illumina sequence data. We would be pleased to test FORENSIC using other technologies, such as minION.
Can I use data generated using another sequencing platform than Illumina?
We only validated the predictions using Illumina sequence data. We would be pleased to test FORENSIC using other technologies, such as minION.
FORENSIC tests automatically for multiples sources, is it possible to test samples for a specific classifier?
The classifiers and the associated predictions are independent. The prediction of one classifier does not influence the predictions of the other classifiers. In other words, identifying the sewage signature in a sample does not weaken the probability of detecting the cow signature in that same sample.
Can I use the voting tree probability as a proxy of fecal contamination percentage?
No! The voting tree probability reflects the similarity between the fecal structure recovered in the tested sample and the signature stored in the classifier. So, it cannot be used to approximate the percentage of contamination in a sample. However, the relationship between these two variables is not linear but logarithmic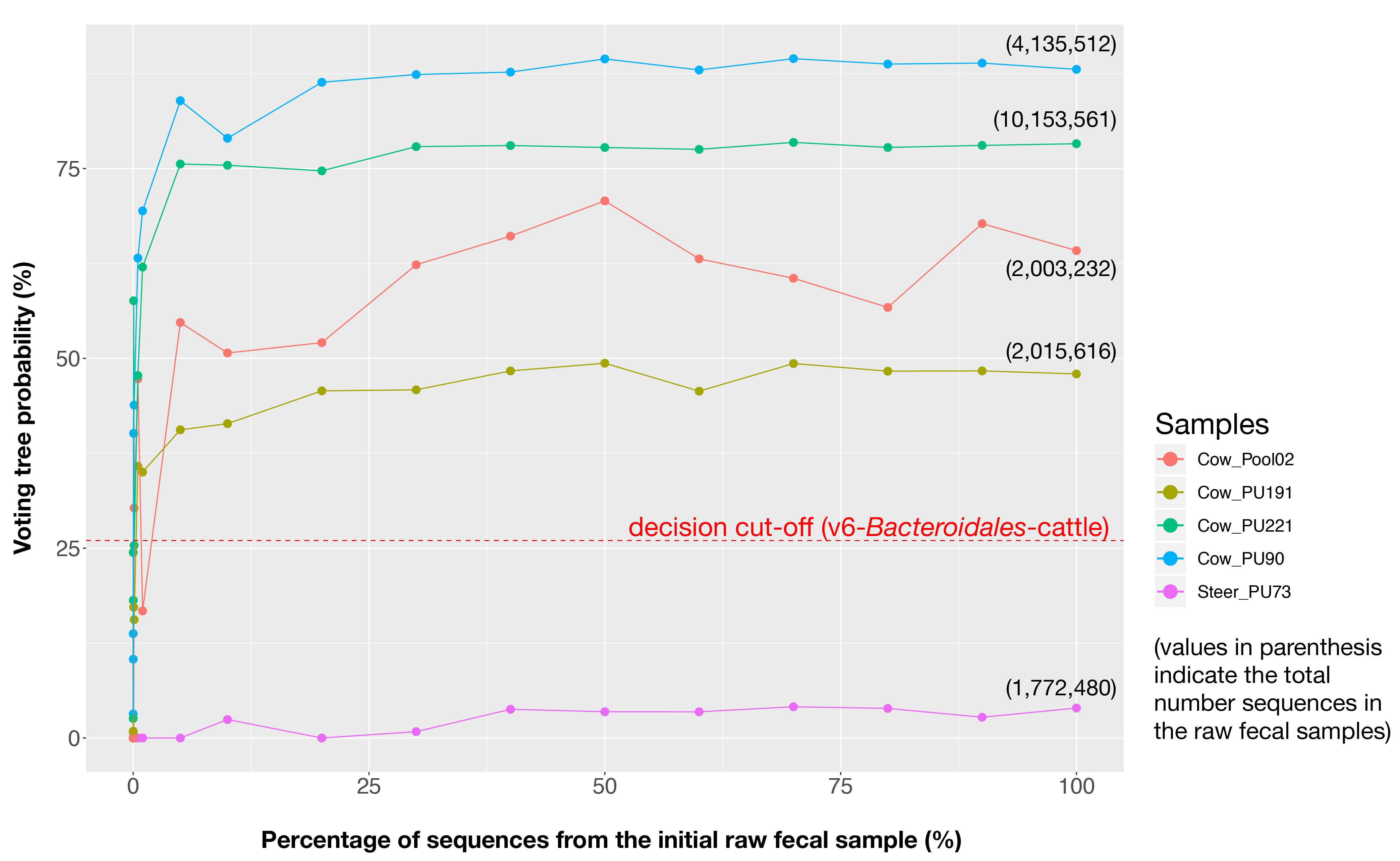 .
.
How long should last an analysis?
The analysis should take a few minutes, depending on the size of your fasta: If it takes longer than that, feel free to contact us.